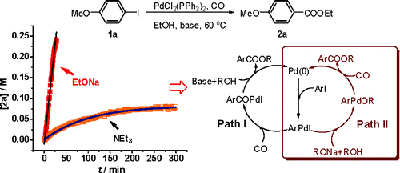
A novel mechanism was proposed for the alkoxycarbonylation of aryl iodides with sodium alkoxide as the base. The catalytic cycle consisted of oxidative addition, subsequent ArPdOR formation, CO insertion to Pd-OR, and final reductive elimination of ArPdCOOR to produce ArCOOR. The kinetic simultaneity of the formation of deiodinated side product from the aryl iodide and aldehyde from corresponding alcohol and the formation of thioether as a competitive product in thiocarbonylation provided strong evidence for the existence of ArPdOR species. Kinetic studies revealed that neither oxidative addition nor reductive elimination was rate limiting and the rate determining step was associated with the reaction of CO with the ArPdOR intermediate. DFT calculations suggested a kinetic preference for CO insertion into Pd-OR bond.
Palladium catalyzed alkoxycarbonylation of organic halides is an important method for fine chemical synthesis. Previously, many mechanistic studies have used tertiary and secondary amines as bases, and the mechanism is generally accepted to consist of oxidative addition of the aryl halide, CO coordination and insertion, and alcoholysis of the resulted acyl palladium complex (Path I). Theoretically, another pathway is possible where the ArPdOR species forms via metathesis from the oxidative addition intermediate ArPdI (Path II).
However, compelling evidence is still demanded for the alternative Path II. The current study toward the Pd-catalyzed alkoxylcarbonylation of aryl iodides with sodium alkoxides as the base revealed a mechanism distinct from Path I and consistent with Path II. Understanding of the mechanism allowed alkoxycarbonylation and thiocarbonylation of aryl iodides to proceed smoothly including the formation of t-butyl esters and thioesters which are usually difficult under classic mechanistic pathway.
The work was carried out be researchers of Wuhan University, Hong Kong University of Science and Technology, Lanzhou Institute of Chemical Physics, CAS. And it was published in the J. AM. CHEM. SOC. (J. AM. CHEM. SOC. 2010, 132, 3153–3158).