The halogen bonding interactions between C6F5I and a series of transition metal monohalides trans-[M(X)(2- C5NF4)-(PR3)2] (M = Ni, Pd, Pt; X = F, Cl, Br; R = Me, Cy) have been studied with quantum chemical calculations.
Optimized geometries of the halogen bonding complexes indicate that angles C1-I···X are basically linear (178-180°) and angles I···X-M mainly range from 90 to 150°.
The strength of these metal-influenced halogen bonds alters with different metal centers, metal-bound halogen atoms and the substitutes on phosphine ligands.
Electrostatic potential and natural bond orbital analysis show that both of the electrostatic and orbital interactions make a contribution to the formation of halogen bonds, while the electrostatic term plays a dominant role.
AIM analysis suggests that, for trans-[M(F)(2- C5NF4)-(PR3)2] (M = Ni, Pd, Pt) monomers, the formed halogen bonding complexes are stabilized by local concentration of the charge of intermediate character, while for the metal monomers containing chlorine and bromine, a typical closed-shell interaction exist.
These results prove that the structures and geometries of these halogen bonding complexes can be tuned by changing the halogen atoms and metal centers, which may provide useful information for the design and synthesis of new functional materials.
Additional Information:
1 Author Information: Na Cheng, Yongjun Liu, Changqiao Zhang, Chengbu Liu Correspondence: yongjunliu_1@sdu.edu.cn.2 Published: J Mol Model , 2013, 19:3821–3829
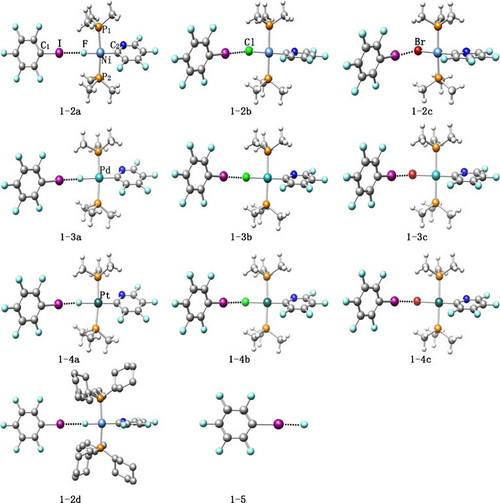
Fig. The optimized structures of halogen-bonding complexes at BhandHLYP/6-31G*-SDD level |
